
固体中の電子間相互作用を厳密に扱うことは非現実的なほど難しいので、密度汎関数法に基づく第一原理計算では、交換相関ポテンシャル(汎関数)を近似して扱います。初期に提案され、現在でも計算負荷が小さいことから頻繁に用いられる汎関数GGAは、様々な材料物性の解析に成功してきた一方で、バンドギャップの過小評価という問題を内包しています。その様子をナローギャップ半導体InSb(バンドギャップ実験値:0.17 eV)で示します。
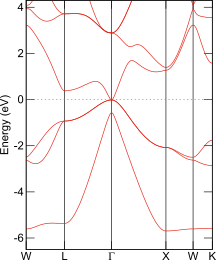
InSbのバンド構造図
PHASE/0; GGA-PBE
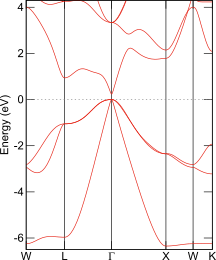
InSbのバンド構造図
PHASE/0; HSE06 + Wannier90
バンドギャップを改善する計算手法の代表がハイブリッド汎関数です。PHASE/0でハイブリッド汎関数HSE06を使うと、InSbはバンドギャップ0.15 eVの半導体となりました。Wannier90と組み合わせて作成したバンド構造図を右に示します。ハイブリッド汎関数を利用すると、バンドギャップ以外にも計算結果の実験値との一致が良くなることが知られていますが、その代償は計算負荷が非常に高くなることです。大規模系に適しているとは言い難い手法です。
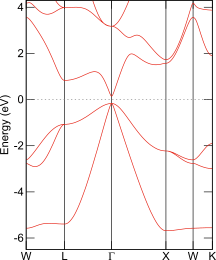
InSbのバンド構造図
Elk; TB09
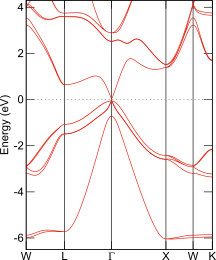
InSbのバンド構造図
Elk; GGA-EV + スピン軌道
そこで、バンドギャップを改善するための簡便な方法として、メタGGAもよく使われます。その例としてTB09でのバンド構造を左図左に示します。バンドギャップの存在が確認できます。ところで、GGAはバンドギャップを過小評価すると述べましたがGGAにも種類があり、あらゆるGGAがInSbのバンドギャップを0と予測するわけではありません。バンド構造の再現に重きを置いた汎関数GGA-EVを使うと、半導体的な電子状態が求まります。スピン軌道相互作用を考慮したバンド構造を左図右に示します。
GGAがバンドギャップを過小評価する度合いは、材料により区々です。ここまでInSbを対象にナローギャップがつぶれる様子を見ましたので、次にワイドギャップが極端に小さくなる例を紹介します。
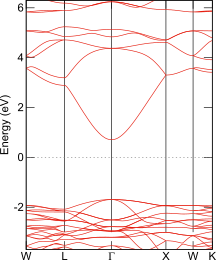
ITOのバンド構造図
Elk; TB09
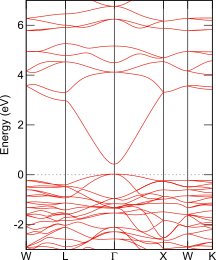
ITOのバンド構造図
ABINIT; GGA-PBEsol
SnO2を添加したIn2O3はITO (Indium Tin Oxide)と呼ばれ、透明電極として利用されますのでそのバンドギャップは、可視光を吸収しない程度に大きな値(> 3 eV)です。SnO2を高濃度に添加したIn2Sn2O7という結晶が知られており、透明電極ほどではないにせよ大きなバンドギャップが期待されます。ところがPBEsolはこれを著しく小さく、ナローギャップ半導体並みの0.4 eVと見積ります。TB09を用いるとバンドギャップは2.4 eVへと大幅に改善します。
現在でもバンドギャップを正確に予測することは、第一原理電子状態計算には荷が重い課題です。添加元素の影響や、表面・界面の電子状態を調べる場合など、バンドギャップを求めること自体が目的でなくとも、その出発点として、母体となる結晶の既知の性質が再現されていることは重要です。弊社受託解析・コンサルティングでは、解析の目的、必要な精度、計算負荷などを考慮して、汎関数の選択を含む適切な計算条件をご提案いたします。
バンド構造図は、逆格子のk点対称線に沿った固有値をプロットして作成しますので、計算結果は逆格子(すなわち実格子)に依存します。論文や教科書に掲載されているバンド構造図は、詳細な情報を含むように最大の逆格子(最小の実格子;基本格子)について描かれます。
擬ポテンシャル法など原子を露わに扱う電子状態計算では、結晶中の不純物を解析する際にスーパーセルを用います。スーパーセルのような大きな実格子では逆格子が小さく折りたたまれるので、通常の計算手順ではバンド構造図も「折りたたまれ」(フォールディングされ)てしまいます。この逆空間で折りたたまれた電子状態を、波動関数の周期性を手掛かりに「解きほぐし」て基本格子に相当するバンド構造図を計算する手法がバンドアンフォールディングです。その活用例をダイヤモンド中の窒素不純物を題材に示します。
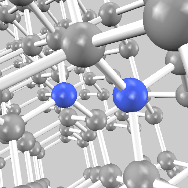
 Ia型CN
Ib型CN
Ia型CN
Ib型CN
宝石を含む天然ダイヤモンドの多くは0.1%程度の窒素を含み(I型)、それら窒素原子は層状に析出することが知られています(Ia型)。一方、単独で分布する窒素原子は合成ダイヤモンドの着色の原因とされ、これを含む構造はIb型に分類されます。両者の性質を調べるために、全250原子の系(基本格子の5x5x5倍スーパーセルに置換型窒素不純物2個)を対象に第一原理計算を行いました(ABINIT利用)。窒素原子がペアを形成した構造をIa型としました(右図)。
安定構造のエネルギーを求めると、Ia型の方がIb型よりも安定であることが分かりました(約3.8 eV)。Ib型天然ダイヤモンドが非常に少ない事実を裏付ける結果です。
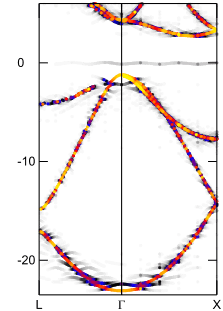
Ia型のバンド構造図
エネルギー (eV)
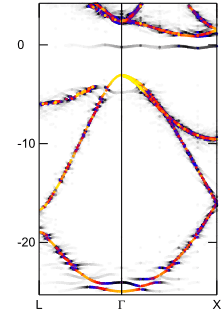
Ib型のバンド構造図
エネルギー (eV)電子状態を詳しく調べるために、Ia型, Ib型それぞれのバンド構造を左図に示します。図中の色は、アンフォールディングの結果得られる「重み」を表します。ダイヤモンド中の窒素原子は「ドナー準位」を形成しますので、その位置(エネルギー原点近傍の灰線)に注目してください。Ia型はドナー準位が価電子帯近くに形成されるのに対して、Ib型では伝導体の近くに形成されます。Ia型では不純物準位が深いので可視光線程度のエネルギーでは電子が励起されず(バンドギャップは過小評価されています)、目で見る限りでは不純物の影響は無さそうです。一方Ib型では浅いドナー準位から伝導体に電子が励起され易く、単独の窒素原子が着色の原因になることが理解できます。
第一原理計算による受託解析を承っております。
電子状態を知りたい、物性値を予測したいなどのご要望、ご納期や費用、またお手元の課題にご対応可能かどうかにつきましても、ぜひ一度お問い合せください。
![]()
![]()
![]()